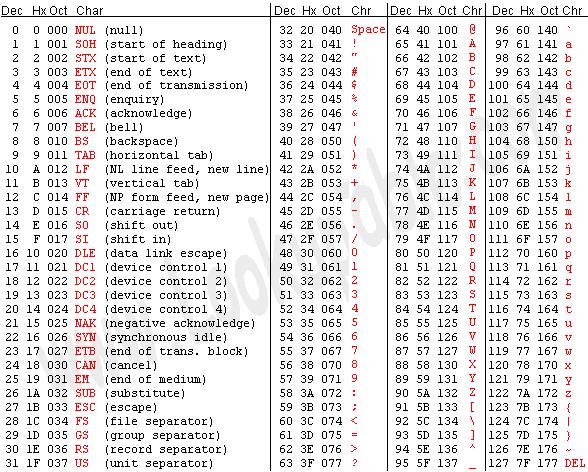
生信常用文件格式
生信常用文件格式
序列格式
fasta
一种基于文本的、用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来表示,且允许在序列前添加序列名及注释,文件名常以
.fasta、.fa结尾。常见后缀说明:
.fasta:普通的FASTA文件 (包括:.fas、 .fasta、.fsa、.fst、.txt和.fa等)
.fna:表示核酸序列的 FASTA 文件
.faa:表示氨基酸序列的 FASTA 文件
.ffn:整个基因组编码区的 FASTA 文件
.frn:以 DNA 字母编码表示的基因组非编码 RNA 区 ( 如tRNA、rRNA ) 的 FASTA 文件格式说明
![Untitled]()
- 每条序列的第一行是由
>开头的任意文字说明。用于序列标记,最好每条序列的标识具有唯一性,一般会用空格把头信息分为两个部分:第一部分是序列名字,它和大于号(>)紧接在一起;第二部分是注释信息,这个可以没有。 - 从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号,其中核苷酸大小写均可,氨基酸只能大写。
- 每条序列的第一行是由
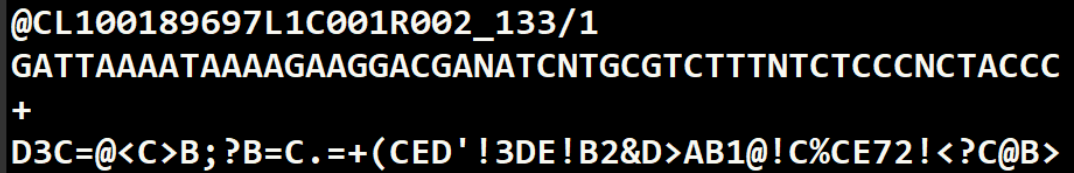
fastq
- 一种基于文本的存储测序生物序列和对应碱基(或氨基酸)质量的文件格式,可以看成fasta文件的变种,一条序列总共包括四个部分。
格式说明
![Untitled]()
![Untitled]()
- 第一行以“@”开头,随后为Illumina 测序标识符 (Sequence Identifiers) 和描述文字 (选择性部分)。
- 第二行是碱基序列。
- 第三行以“+”开头,随后为Illumina 测序标识符 (选择性部分)。
第四行是对应碱基的测序质量,该行中每个字符对应的 ASCII 值减去 33,即为对应第二行碱基的测序质量值。
![Untitled]()
常用命令
1
2
3echo "`CL100189782_L01_read_1.fq.gz | wc -l` / (4*1000000)" | bc -l
# 测序碱基数计算
zcat trt_N061011_1.fq.gz | awk'{if(FNR%4==0) base+=length}END{print base/10^9,"G";}'

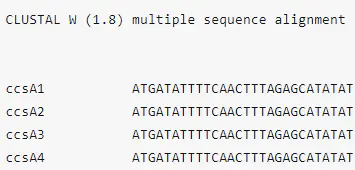
序列比对格式
fasta
- 最常见的多序列比对格式,和序列的fasta格式类似,只是为了使整体出现最大的可能性,在序列中可能会添加一些
-。 格式说明
![Untitled]()
- 最常见的多序列比对格式,和序列的fasta格式类似,只是为了使整体出现最大的可能性,在序列中可能会添加一些
clustal
- clustal格式的文件是纯文本格式,它可以选择有一个头来声明clustal版本号。接下来是多序列比对,以及关于比对中每个位置保存程度的可选信息。
格式说明
![Untitled]()
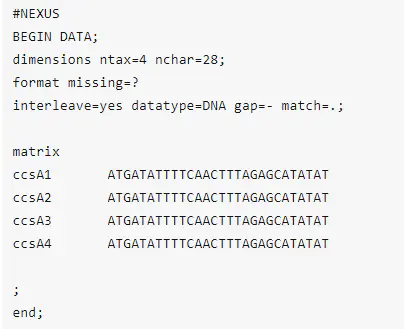
NEXUS
- 一种文本格式,使用“块”的方式来组织信息。以#NEXUS开头,后续说明了总体信息(包括序列数量,大小,数据类型,缺失数据等
格式说明
![Untitled]()
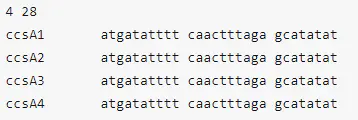
PHYLIP
- 主要包括两个部分:一个标题描述对齐维度(序列数量和大小),后跟多序列对齐序列。
格式说明
![Untitled]()

常见文件格式
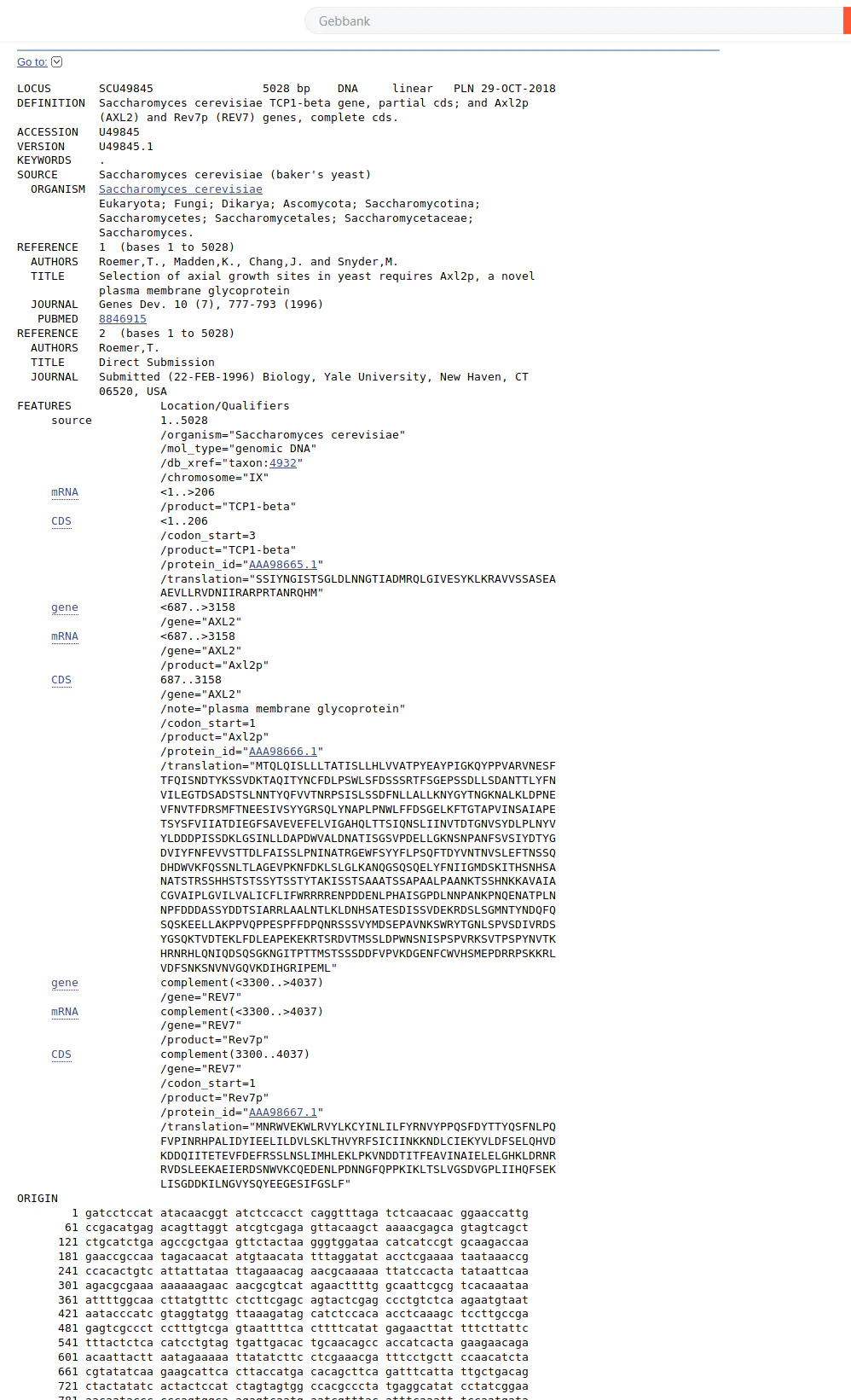
Genbank
- 最早的生物信息学数据格式之一,包含的信息十分全面,常以
.gbff结尾 格式说明
![Untitled]()
- 最早的生物信息学数据格式之一,包含的信息十分全面,常以
GFF和GTF
GFF (General Feature Format) 和 GTF (Gene Transfer Format) 都是用于存储注释信息的文本类型。目前常用GFF格式为第二版本的GFF2和第三版本的GFF3,GTF常用GTF2。两者前8列是相同的,GTF格式相交GFF格式更加严格。以GFF3格式为例进行说明。
常用命令
1
2# grep 匹配查询 -v 输出不匹配的行
gunzip Homo_sapiens.GRCh38.94.gtf.gz -c |grep -v '^#' | sed '/^[^chr]/ s/^/chr/' >GRCh38.gtf格式说明
1
2
3
4
5NZ_CP068034.2 RefSeq region 1 6018586 . + . ID=NZ_CP068034.2:1..6018586;Dbxref=taxon:317;Is_circular=true;Name=ANONYMOUS;collection-date=2001-05-30;country=Belarus: Minsk region;gbkey=Src;genome=chromosome;isolation-source=Ribes nigrum leaves;lat-lon=53.893009 N 27.567444 E;mol_type=genomic DNA;nat-host=Ribes nigrum;strain=BIM B-268
NZ_CP068034.2 RefSeq gene 1 1536 . + . ID=gene-JJQ97_RS00005;Dbxref=GeneID:64441043;Name=dnaA;gbkey=Gene;gene=dnaA;gene_biotype=protein_coding;locus_tag=JJQ97_RS00005;old_locus_tag=JJQ97_25475
NZ_CP068034.2 Protein Homology CDS 1 1536 . + 0 ID=cds-WP_003437057.1;Parent=gene-JJQ97_RS00005;Dbxref=Genbank:WP_003437057.1,GeneID:64441043;Name=WP_003437057.1;gbkey=CDS;gene=dnaA;inference=COORDINATES: similar to AA sequence:RefSeq:NP_064721.1;locus_tag=JJQ97_RS00005;product=chromosomal replication initiator protein DnaA;protein_id=WP_003437057.1;transl_table=11
NZ_CP068034.2 RefSeq gene 1575 2678 . + . ID=gene-JJQ97_RS00010;Dbxref=GeneID:64441044;Name=dnaN;gbkey=Gene;gene=dnaN;gene_biotype=protein_coding;locus_tag=JJQ97_RS00010;old_locus_tag=JJQ97_25480
NZ_CP068034.2 Protein Homology CDS 1575 2678 . + 0 ID=cds-WP_201418908.1;Parent=gene-JJQ97_RS00010;Dbxref=Genbank:WP_201418908.1,GeneID:64441044;Name=WP_201418908.1;gbkey=CDS;gene=dnaN;inference=COORDINATES: similar to AA sequence:RefSeq:NP_064722.1;locus_tag=JJQ97_RS00010;product=DNA polymerase III subunit beta;protein_id=WP_201418908.1;transl_table=111
2
3
4
5
6
7
8seqname:序列定位,必须为染色体或组装好的
scaffoldsource:产生该GTF/GFF 文件的项目名称
feature:该段序列的特征:如 exon,CDS,enhancer等
start:序列起始位点
end:序列终止位点
score:得分值,如无得分,则为“.”
strand:正负链,用“+”和“-”表示,如不关心或者缺乏正负链信息,则为“.”
frame:开放阅读框,分别用0,1,2来区别,如果是非编码序列,则为“.”相同点
feature:GTF的feature type受限于使用软件的规定,GFF的feature可以为任意内容。score:GTF的score一般不会被用到,都是“.”
attribute:GTF的第九列为attribute,为序列对应的属性,其中的内容包括序列对应的 gene_id 和 transcript_id,一般还有序列中包含的外显子数量,在GFF3版本中第九列也为attribute,但不同属性中用“=”相隔,GTF格式中不同属性用“;”分隔
group:GFF2的第九列为group,来自同一个组的不同序列都被具有相同的组名。
BED
- 分析过程中的bed文件一般代表区域信息
区别
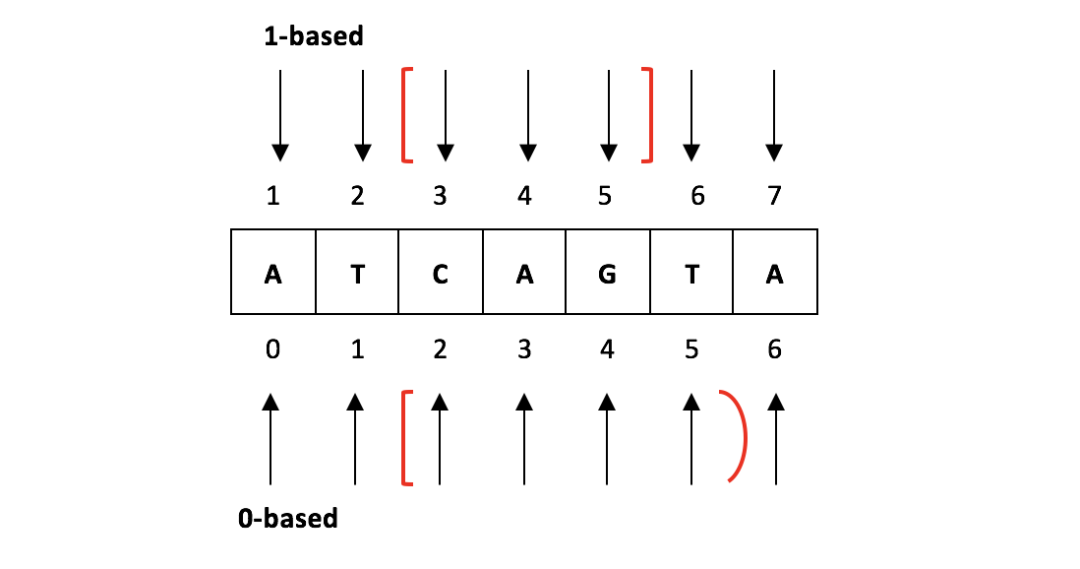
表示基因注释时,gtf/gff和bed文件的区别
- gtf/gff文件一行表示一个exon/CDS等子区域,多行联合表示一个gene;bed文件一行表示一个gene
gtf文件中碱基位置定位方式是1-based,而bed中碱基定位方式是0-based
![Untitled]()
格式说明
- 必须包含的3列信息:
- chrom:染色体名字 (e.g.chr3, chrY, chr2_random或者scaffold10671)
- chromStart:基因在染色体或scaffold上的起始位置(0-based)
- chromEnd:基因在染色体或scaffold上的终止位置 (前闭后开)
- 可选的9列信息:
- name:bed文件的行名
- score:本条基因在注释数据集文件中的评分(0-1000),在Genome Browser中会根据不同区段的评分显示对应的阴影强度(评分越高灰度越高)
- strand:链的方向
+、-或.(.表示不确定链的方向) - thickStart:CDS区(编码区)的起始位置,即起始密码子的位置。
- thickEnd:The endingposition at which the feature is drawn thickly (for example the stop codon ingene displays).
- itemRgb:RGB颜色值(如:255,0,0),方便在GenomeBrowser中查看
- blockCount:bed行中外显子的数目
- blockSizes:
逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的碱基数。 - blockStarts:
逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的起始位置(数值是相对ChromStart计算的)。
- 必须包含的3列信息:
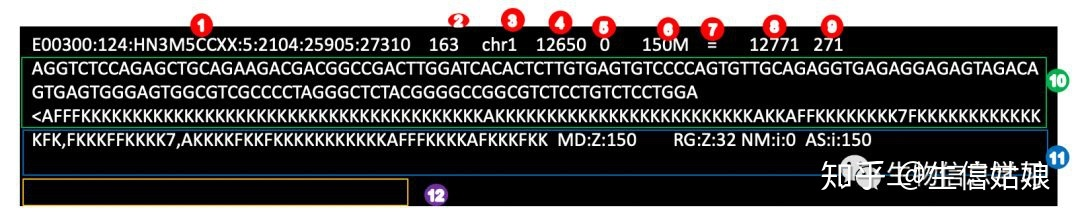
SAM
Alignment/Map步骤bwa/STAR/HISAT2等软件对结果的标准输出文件,用于存储reads比对到参考基因组的比对结果,是一个纯文本格式,文件一般较大。为了节省硬盘存储,一般使用其高效压缩的二进制格式
bam文件。1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17# bam文件查看方式
samtools view filename.bam | less -S
samtools view -h filename.bam | less -S
###结果展示
@HD VN:1.5 SO:coordinate
@SQ SN:chr1 LN:248956422
@SQ SN:chr10 LN:133797422
......
@SQ SN:chrKI270392.1 LN:971
@SQ SN:chrKI270394.1 LN:970
@RG ID:BH_H3K27ac_2 LB:BH_H3K27ac_2 SM:BH_H3K27ac_2
@PG ID:bwa PN:bwa VN:0.7.15-r1140 CL:bwa mem -M -t 8 -R@RG\tID:BH_H3K27ac_2\tLB:BH_H3K27ac_2\tSM:BH_H3K27ac_2\tPL: /MP
@PG ID:MarkDuplicates VN:1.138(aa51703435dc6a423013e74e56b0b68405facd79_1439324166) CL:picard.sam.markduplicates.
K00141:244:HVL3NBBXX:8:2119:27235:3145399 chr1 10016 32 115M = 10016 115 CCCTAACCCTAACCCTAACCC
K00141:244:HVL3NBBXX:8:2119:27235:31453147 chr1 10016 32 115M = 10016 -115 CCCTTACCCTAACCCTAACCC格式说明
- header内容
- @HD:是必须的标准文件头,包含版本信息;
- @SQ:参考序列染色体名字和长度信息 (SN:scaffold name; LN: length);
- @RG:重要read group信息,通常包含测序平台,测序文库和样本ID等信息,分析时用于区分不同样本(重测序时用到);
- @PG:生成此文件的操作过程和参数信息 (program)
record内容
1
2
3
4
5
6
7
8
9
10
11
121. read名称;
2. 比对信息位flag值;
3. 参考序列染色体编号;
4. 5′端起始位置;
5. MAPQ:mapping quality,描述比对的质量,数字越大,特异性越高;
6. CIGAR字符串,记录插入、删除、错配等信息;
7. 配对read所比对到的染色体,仅双端测序的数据才有;
8. 配对read所比对到的位置,仅双端测序的数据才有;
9. 插入片段的长度,仅双端测序的数据才有;
10. read序列;
11. read质量值;
12. 12列以后的信息都是metadata,程序用标记![Untitled]()
- header内容
BAM